This article “Amorphous Solid Dispersions Layered onto Pellets – An Alternative to Spray Drying?” is an excerpt from the publication of Neuwirth et al., Pharmaceutics 2023, 15(3), 764; https://doi.org/10.3390/pharmaceutics15030764.
Abstract
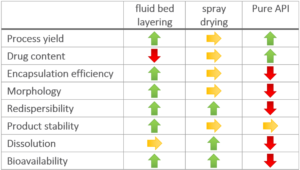
Spray drying is one of the most frequently used solvent-based processes for manufacturing amorphous solid dispersions (ASDs). However, the resulting fine powders usually require further downstream processing when intended for solid oral dosage forms. In this study, we compare properties and performance of spray-dried ASDs with ASDs coated onto neutral starter pellets in mini-scale. We successfully prepared binary ASDs with a drug load of 20% Ketoconazole (KCZ) or Loratadine (LRD) as weakly basic model drugs and hydroxypropyl-methyl-cellulose acetate succinate or methacrylic acid ethacrylate copolymer as pH-dependent soluble polymers. All KCZ/ and LRD/polymer mixtures formed single-phased ASDs, as indicated by differential scanning calorimetry, X-ray powder diffraction and infrared spectroscopy. All ASDs showed physical stability for 6 months at 25 °C/65% rH and 40 °C/0% rH. Normalized to their initial surface area available to the dissolution medium, all ASDs showed a linear relationship of surface area and solubility enhancement, both in terms of supersaturation of solubility and initial dissolution rate, regardless of the manufacturing process. With similar performance and stability, processing of ASD pellets showed the advantages of a superior yield (>98%), ready to use for subsequent processing into multiple unit pellet systems. Therefore, ASD-layered pellets are an attractive alternative in ASD-formulation, especially in early formulation development at limited availability of drug substance.
Materials
The model drugs ketoconazole (KCZ) and loratadine (LRD) were purchased from Sris Pharmaceuticals (Hyderabad, India). HPMCAS LG (hydroxypropyl-methylcellulose acetate succinate, wt%: methoxyl 20–24%, hydroxypropyl 5–9%, succinyl 14–18%; Mw = 18,000, HPMC-AS) was donated from Shin-Etsu Chemical (Tokyo, Japan). Eudragit L100-55 (methacrylic acid ethylacrylate copolymer, ratio 1:1, Mw = 320,000, EL100-55) was donated by Evonik (Darmstadt, Germany). Cellets 1000 (microcrystalline cellulose starter pellets, 1000–1400 µm) were provided by Glatt Pharmaceutical Services (Binzen, Germany). A detailed list of the pellets’ characteristics is shown in Table 1. Ethanol 96% (v/v) (technical grade) used in the sample preparation, and methanol (analytical grade) used for the HPLC analytics as well as the buffer salts disodium mono-hydrogen phosphate dodecahydrate (Na2HPO4·12H2O) and monosodium dihydrogen phosphate dodecahydrate (NaH2PO4·12H2O) were obtained from VWR Chemicals GmbH (Darmstadt, Germany).
| Pellet Properties | |
|---|---|
| d50 (xc min) [µm] | 1123.44 (±7.36) |
| SPAN | 0.166 (±0.002) |
| b/l | 0.893 (±0.000) |
| SPHT | 0.956 (±0.001) |
| Particle density [g/cm3] | 1.452 (±0.016) |
| Sm [cm2/g] | 36.41 (±0.29) |
Table 1. Pellet properties of Cellets 1000. d50: mean particle diameter determined by the particle width; SPAN: width of the particle distribution; b/l: aspect ratio; SPHT: sphericity; Sm: specific surface area.
Pellet Coating (PC)
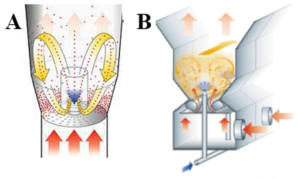
For pellet coating (PC) a laboratory scale fluid bed system Mini Glatt equipped with a Micro-Kit (Glatt GmbH, Binzen, Germany) was used. The coating was applied with a 0.5 mm two-fluid nozzle in bottom spray using the special bottom plate of the Micro-Kit to emulate a three-fluid nozzle with micro-climate. In the beginning, the machine was filled with 25.0 g of Cellets® 1000. The following process parameters were maintained throughout the process: Process gas flow 30 m3/h, product temperature 30.0 ± 1.0 °C (resulting inlet temperature 32–35 °C), spray pressure 1.5 bar and spray rate 1.0 ± 0.2 g/min. The final pellets had a theoretical drug-load of 10% (w/w) due to the fact that ASD and core pellets were used in a 1:1-ratio. […] To prepare the spraying solutions, the API and polymer were dissolved in ethanol 96% (v/v) under continuous stirring (solid content of 10% (w/w)). Prior to spraying, each solution was sonicated for 15 min to ensure complete dissolution of the components.
Subsequently, the coated pellets were manually sieved with a 2 mm mesh to eliminate multicore pellets. The pellets were dried under vacuum for 24 h at the same conditions as the SD powder.
Conclusions
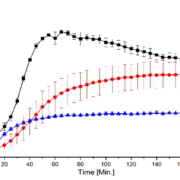
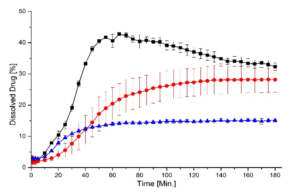
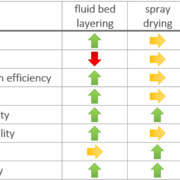
In this study, we successfully prepared binary single phase ASDs of KCZ and LRD as weakly basic, slow crystallizing model APIs (drug load 20% (w/w)) using HPMC-AS or EL100-55 as pH-dependent soluble polymers via fluid bed pellet coating and spray drying. While the received ASD-pellets would not require further downstream processing other than capsule filling or tableting, the fine SD powder had to be transformed into dry granules. In combination with the slow crystallizers, KTZ and LOR, both manufacturing processes resulted in single-phased ASDs of high physical stability (up to 6 months) and similar dissolution performance when normalized to the total outer surface. The dissolution rate depends mainly on this total outer particle surface of the respective sample, independent of the manufacturing process, while the porosity of the sample had a minor impact on its dissolution behavior.
Especially for early formulation development, the high yield and ease of handling due to the pellet properties are strong advantages over the standard spray drying process. Nevertheless, the long process time in larger scale requires further process optimization in fluidized bed processing.

Figure: Amorphous solid dispersions.
[1] Neuwirth et al., Pharmaceutics 2023, 15(3), 764; https://doi.org/10.3390/pharmaceutics15030764
 Ingredient Pharm
Ingredient Pharm